Beckwith-Wiedemann syndrome
Beckwith-Wiedemann syndrome is a genetic condition caused by disruption of the chromosome region 11p15 genomic imprinting.
Overview
Beckwith-Wiedemann syndrome (BWS) is characterised by prenatal overgrowth, macroglossia, exomphalos, lateralised overgrowth and a predisposition to the development of embryonal tumours (particularly Wilms tumour).
Clinical features
Cardinal and suggestive features of BWS are listed below. According to consensus criteria published in 2018, which include a scoring system, a clinical diagnosis of BWS should be made (even in the absence of a confirmatory 11p15 result) when an individual scores at least four points (the cardinal features listed below attract a score of two and the suggesting features attract a score of one).
Cardinal features of BWS (two points per feature)
The cardinal features of BWS are:
- macroglossia;
- exomphalos;
- lateralised overgrowth (hemihypertropy);
- multi-focal and/or bilateral Wilms tumour or nephroblastomatosis;
- hyperinsulinism (lasts beyond the first week of life); and
- pathology: adrenal cortex cytomegaly, placental mesenchymal dysplasia, pancreatic adenomatosis.
Suggestive features of BWS (one point per feature)
Suggestive features of BWS are:
- birth weight more than two standard deviations (2SD) above the mean (equivalent to the 98th percentile as described in the UK-World Health Organization growth charts);
- facial naevus flammeus;
- polyhydramnios and/or placentomegaly;
- anterior ear lobe creases and/or posterior helical pits;
- transient hypoglycaemia (resolves within the first week of life);
- BWS-associated tumour (not bilateral or multi-focal Wilms tumour), unilateral Wilms tumour, hepatoblastoma, neuroblastoma, rhabdomyosarcoma, adrenocortical carcinoma and/or phaeochromocytoma;
- nephromegaly and/or hepatomegaly; and
- umbilical hernia and/or diastasis recti.
Clinical details
- Macroglossia: a large tongue can cause feeding difficulties, impact upon breathing and sometimes interfere with speech. Tongue reduction surgery (at a specialist centre) may be considered.
- Lateralized overgrowth (sometimes called hemihypertrophy or hemihyperplasia): one side of the body (limbs and/or face) is larger than the other.
- Embryonal tumours: BWS is associated with an increased susceptibility to developing embryonal tumours. The most common is Wilms tumour. Nephroblastomatosis is the precursor to Wilms tumour. The embryonal tumour risk is related to the underlying 11p15 epigenotype (table 1).
- Macrosomia: high birth weight, reflective of prenatal overgrowth, which can lead to premature and/or problematic labour. Growth often normalises as an individual gets older.
- Anterior abdominal wall anomalies: a range of abdominal wall anomalies are associated with BWS. They include exomphalos (also known as omphalocele, and in which the abdominal contents, covered by peritoneal membrane, protrude externally), umbilical hernia and diastasis recti (weakness of the anterior abdominal wall muscles, which results in their separation).
- Abdominal organomegaly: where there is lateralised overgrowth, the organs on this side of the body will often be larger than the counterparts on the other side (known as unilateral organomegaly).
- Neonatal hypoglycaemia: low blood sugar due to high insulin production by the pancreas.
- Ear lobe creases and/or pits: the creases are usually located on the anterior ear lobes (it can appear as though the ear has been pierced), and the pits are usually located on the posterior helix.
- Facial nevus flammeus: a flat capillary malformation on the face and/or eyelids, frequently in the glabellar region.
The diagnosis of BWS is achieved through a combination of identification of clinical features, DNA methylation studies, DNA sequencing and/or microarray studies.
Genetics
BWS is caused by disruption of the 11p15 genomic imprinting. This imprinting is controlled by two imprinting centres – imprinting centre 1 (IC1) and imprinting centre 2 (IC2) – which, using methylation ‘switches’, maintain a balance between genes that are paternally expressed and promote growth and genes that are maternally expressed and suppress growth (figure 1).
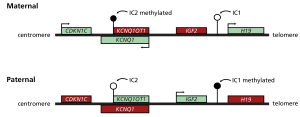
Figure 1: Normal expression pattern for genes in the 11p15 region in unaffected individuals.
11p15 growth genes are differentially expressed dependent upon parent of origin. This differential expression profile is maintained by two imprinting centres: the telomeric imprinting centre 1 (IC1) and centromeric imprinting centre 2 (IC2).
This figure shows the normal expression pattern for the genes in this region, in unaffected individuals. Filled lollipops indicate methylated imprinting centres and open lollipops indicate unmethylated imprinting centres. Arrows indicate transcription. In unaffected individuals, the maternal IC1 is unmethylated, H19 is expressed and IGF2 is silenced. Conversely, the paternal IC1 is methylated, H19 is silenced and IGF2 is expressed. The maternal IC2 is methylated, KCNQ1OT1 is not expressed and CDKN1C and KCNQ1 are expressed. Conversely, neither CDKN1C nor KCNQ1 are expressed from the unmethylated IC2 paternal allele, but the KCNQ1OT1 antisense RNA is.
In individuals with BWS, however, this pattern of gene expression is disrupted. The balance between maternally and paternally expressed genes is disrupted in the following ways:
- loss of methylation at IC2 on the maternal chromosome, impacting growth suppression;
- gain of methylation at IC1 on the maternal chromosome, impacting growth promotion;
- paternal uniparental disomy (UPD) (where both copies of chromosome 11 are from the father); and
- pathogenic variant in the maternal copy of CDKN1C.
The contribution of each of the epigenotypes to BWS is shown in table 1.
For information about testing, see ‘Child with suspected Beckwith-Wiedemann syndrome’ and ‘Genomic imprinting’.
Inheritance and genetic counselling
Most individuals with BWS are the only affected members of their family, though there are rare exceptions.
- Most individuals with BWS are unlikely to have children with BWS (offspring risk), and most unaffected parents of children with BWS are unlikely to have another affected child (recurrence risk).
- The exception to this is if a female (mother or patient) has a CDKN1C gene variant or there is a maternal imprinting centre heritable alteration (such as a deletion). In both of these instances, the offspring/recurrence risk is 50%.
- There are also rare instances of chromosome rearrangements that are associated with an increased offspring and recurrence risk.
- The recurrence risk of BWS is complex. It is thought that in those patients in whom it is a consequence of loss or gain in methylation, and no microdeletions or microduplications are found (about 85% of cases), the risk is less than 1%. However, for those patients who have a single-gene variant driving pathogenesis, the inheritance can be as high as 50% depending on the parental genotypes. Due to these complexities, you should refer the patient and their family to your local clinical genetics service for a thorough discussion.
The underlying epigenotype also has implications for counselling regarding tumour risk, as shown below in table 1.
| Epigenotype | Contribution to BWS | Tumour risk | Predominant tumour types |
|---|---|---|---|
| IC2 loss of methylation | 50% | 2.6% | Wilms tumour risk is low. Patient is more likely to develop hepatoblastoma, rhabdomyosarcoma and/or neuroblastoma. |
| Paternal UPD 11 | 20% | 16% | Wilms tumour, hepatoblastoma, rhabdomyosarcoma and neuroblastoma. |
| IC1 gain of methylation | 5% | 28% | Wilms tumour. |
| CDKN1C variant | 5% | 6.9% | Wilms tumour risk is low. Patient is more likely to develop hepatoblastoma, rhabdomyosarcoma and/or neuroblastoma. |
Data shown in this table is from ‘Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement’.
Management
Management of children with BWS is complex and should be delivered via a multidisciplinary team following consensus guidelines – including tumour screening in young children, which is a key component of the long-term follow-up of children with BWS.
Resources
For clinicians
- Genomics England: NHS Genomic Medicine Service (GMS) Signed Off Panels Resource
- National Organization for Rare Disorders (NORD): Beckwith-Wiedemann syndrome
- NHS England: National Genomic Test Directory
- Online Mendelian Inheritance in Man (OMIM): Beckwith-Wiedemann syndrome
- Orphanet: Beckwith-Wiedemann syndrome
- US National Library of Medicine: ClinicalTrials.gov database
References:
- Beckwith JB, Shuman C and Weksberg R. ‘Beckwith-Wiedemann syndrome’. European Journal of Human Genetics 2009: volume 18, pages 8–14. DOI: doi.org/10.1038/ejhg.2009.106
- Brioude F, Kalish JM, Mussa A and others. ‘Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement’. Nature Reviews Endocrinology 2018: volume 14, pages 229–249. DOI: 10.1038/nrendo.2017.166
For patients
- Great Ormond Street Hospital for Children NHS Foundation Trust: Beckwith-Wiedemann syndrome (BWS)