Congenital adrenal hyperplasia
Congenital adrenal hyperplasia is an inherited condition where the adrenal gland is larger than usual. Over 90% of cases are caused by a pathogenic variant in the CYP21A2 gene.
Overview
Congenital adrenal hyperplasia (CAH) is an autosomal recessive condition caused by single gene anomalies in the enzymes involved in cortisol biosynthesis, most commonly 21-hydroxylase. This can lead to an inability of the body to cope with physiological stress and lead to virilisation.
Clinical features
The clinical presentation of CAH reflects the involvement of the adrenal glands in the production of:
- mineralocorticoids (aldosterone);
- cortisol; and
- androgens (testosterone).
Each of the CAH conditions results from a defect in one or more of the enzymes that are responsible for adrenal hormone synthesis (see figure 1). Where there is a defect, this will result in a reduction in the hormone downstream of that defect and an increase in other hormones upstream.
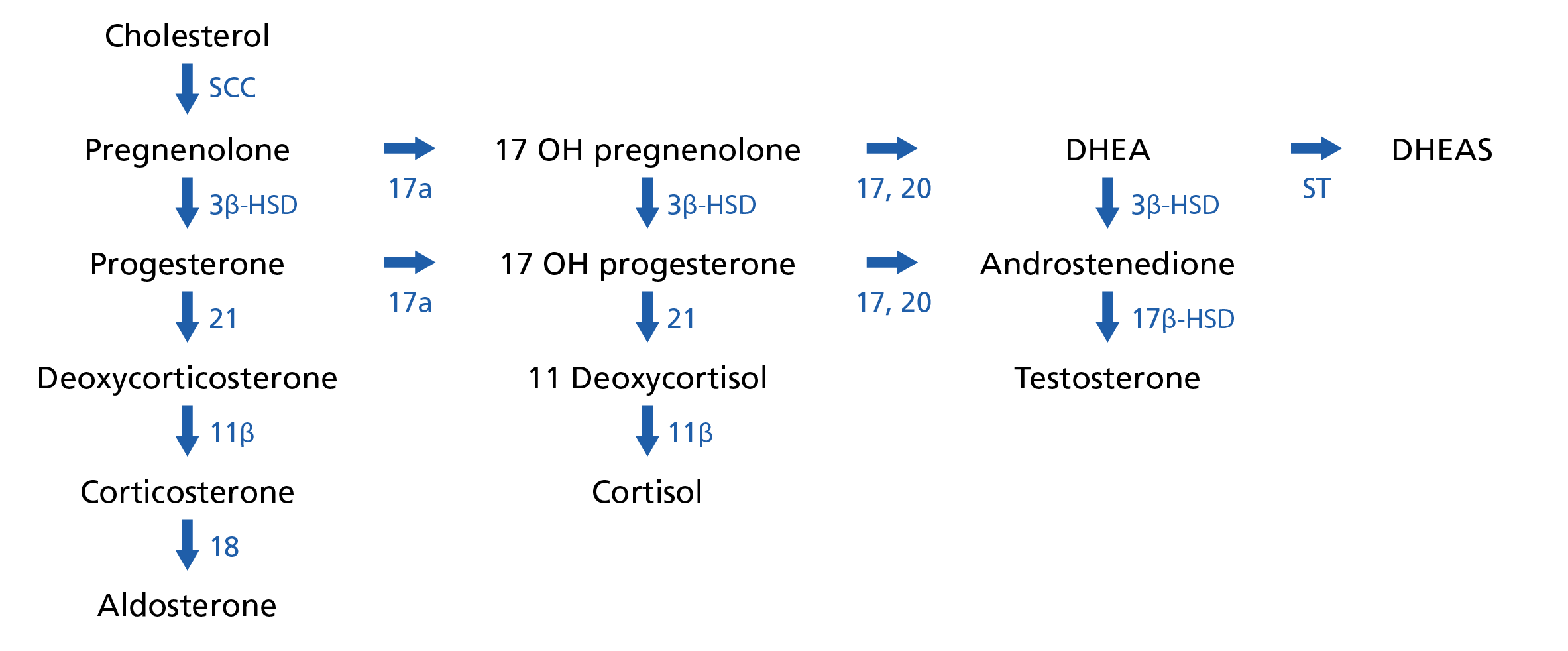
Figure 1: Steroid synthesis pathways
Figure legend: SCC, cholesterol side-chain cleavage enzyme; 3β-HSD, 3β-hydroxysteroid dehydrogenase; 17a, 17α-hydroxylase; 17,20, 17,20 lyase; ST sulfotransferase; 21, 21-hydroxylase; 17β-HSD, 17β-hydroxysteroid dehydrogenase; 11β, 11β-hydroxylase; 18, 18-hydroxylase.
Diagram adapted from WikiMedia Commons: Adrenal steroid hormone synthesis. Licensed under the Creative Commons Attribution-Share Alike 4.0 International license.
{kind=link}
The most common form of CAH, responsible for over 90% of cases, is caused by a pathogenic variant in the gene encoding the 21-hydroxylase enzyme (CYP21A2). This causes a reduction in aldosterone and cortisol but an increased production of testosterone, as the aldosterone and cortisol metabolites are pushed down this branch of the pathway.
There are two forms of 21-hydroxylase deficient CAH: classical and non-classical.
Classical 21-hydroxylase deficient CAH
75% of children with classical CAH have a salt wasting (SW) form. Inadequate aldosterone production results in insufficient sodium reabsorption from the distal renal tubules. Initially children present with failure to thrive, poor feeding, vomiting, dehydration, hypotension, hypoglycaemia, hyperkalaemia, and hyponatraemia. If untreated, children will progress to a life-threatening adrenal crisis with hyperkalaemic metabolic acidosis.
25% of children with classical CAH have a simple virilising (SV) form. In classical virilising CAH, newborns are exposed to excess androgens. This results in virilisation of 46,XX females, who develop normal internal female organs, but may be born with ambiguous or male external genitalia, which may be identified at birth, or during the newborn and infant physical examination (NIPE). Untreated males and females with SV-CAH develop signs of androgen excess including precocious development of pubic hair, increased linear growth, acne, shorter adult height and infertility. The incidence of classical CAH is 1 in 7000–15,000.
Non-classical 21-hydroxylase deficient CAH
Individuals with non-classical CAH (NC-CAH) are usually mildly affected and present later in childhood, often with signs of hyperandrogenism. Individuals with NC-CAH may have early puberty and infertility.
Summary of forms of CAH
Table 1 summarises the various forms of CAH, including the related gene variant, and whether there is an excess or deficiency of androgen and mineralocorticoid.
Table 1: CAH forms and related gene variants
| Form of CAH | Gene | Androgen | Mineralcorticoid |
| Most common form of CAH | |||
| 21-hydroxylase deficiency | CYP21A2 | Excess | Deficient |
| Rare forms of CAH | |||
| 11β-hydroxylase deficiency | CYP11B1 | Excess | Excess |
| 17α-hydroxylase deficiency | CYP17A1 | Deficient | Excess |
| 3β-hydroxysteroid dehydrogenase deficiency | HSD3B2 | Deficient | Deficient |
| Congenital lipoid adrenal hyperplasia | STAR | Deficient | Deficient |
| Cytochrome p450 oxidoreductase deficiency | POR | Deficient | Excess (mild) |
Gene(s)
21-hydroxylase deficient CAH is caused by pathogenic variants in both copies (biallelic) of the CYP21A2 gene, which either inactivate or significantly impair its function. In most patients, each of these variants are different; this is termed “compound heterozygosity”.
While there are over 200 disease-causing CYP21A2 variants, the 10 most common variants account for around 70% of cases. Genotype–phenotype correlations can sometimes be used to predict disease severity and ensure accurate genomic counselling. Rarer forms of CAH involve biallelic changes in CYP11B1, CYP17A1, HSD3B2, STAR and POR.
For information about testing see: Presentation: Child with suspected congenital adrenal hyperplasia, Presentation: Pregnancy at risk of congenital adrenal hyperplasia and Presentation: Fetus with ambiguous genitalia.
Inheritance and genomic counselling
CAH is an autosomal recessive condition that affects males and females in equal numbers. The parents of most affected individuals are carriers for the condition and therefore have a 25% (1-in-4) chance of another child being affected. In the UK, approximately 1-in-50 individuals carry a CAH gene variant. Genomic counselling is particularly important so that any future pregnancies can be managed appropriately.
Preimplantation genomic testing for monogenic conditions (PGT-M) and prenatal testing is available where the familial pathogenic variant is known.
Management
Prompt diagnosis is crucial to initiate appropriate therapy with glucocorticoids and/or mineralocorticoids, and to stop the effects of their deficiency. Individuals with ambiguous genitalia need to be seen by a multidisciplinary team, with the input of specialists in paediatric endocrinology, paediatric urology/surgery, clinical genetics and clinical psychology. It is important that the community midwife, health visitor and GP are informed of care and treatment.
In all forms of the condition, health, fertility and lifespan can be restored to normal with appropriate hormone replacement therapy. Patients with CAH should wear or carry a medical alert identification specifying adrenal insufficiency.
Management of CAH during pregnancy
- This ideally starts with preconception education and genomic counselling. If an at-risk couple wish to avoid having a child affected by CAH, in vitro fertilisation (IVF) with preimplantation genomic testing for monogenic conditions (PGT-M) be undertaken.
- Prenatal screening and diagnostic options include:
- fetal sexing in early pregnancy (from 7–9 weeks) via cell-free fetal DNA from maternal plasma (See: Non-invasive prenatal diagnosis (NIPD)); and
- diagnosis via chorionic villus sampling (CVS) (from 11 weeks) and amniocentesis (from 15 weeks).
- NIPD samples taken between 7 and 9 weeks gestation may require a second confirmatory sample.
- NIPD can also be a direct confirmatory test without the need for CVS or amniocentesis, provided the necessary parental and affected sibling samples have been obtained and validation work has been performed, hence the importance of pre-pregnancy counselling. For further details see Presentation: Pregnancy at risk of congenital adrenal hyperplasia.
- Prenatal dexamethasone (Pdex) treatment, when commenced by the mother before 8 weeks gestation, can prevent virilisation in female fetuses affected by CAH.
- Fetal sexing by NIPD allows Pdex treatment to be stopped in pregnancies that are predicted to be male.
- Pdex treatment is controversial and the 2018 Endocrine Society Clinical Practice Guidelines recommend that clinicians continue to regard prenatal therapy as experimental.
Newborn screening
Based on the last UK National Screening Committee review (November 2021), newborn screening for CAH is not currently recommended.
Resources
For clinicians
- Genomics England: NHS Genomic Medicine Service (GMS) Signed OFF Panels Resource
- National Organisation for Rare Disorders (NORD): Congenital Adrenal Hyperplasia
- NHS England: National Genomic Test Directory
- Public Health England: Newborn and infant physical examination (NIPE) screening programme handbook
References:
- Ahmed SF, Achermann J, Alderson J and others. ‘Society for Endocrinology UK Guidance on the initial evaluation of a suspected difference or disorder of sex development (Revised 2021)’. Wiley Online Library 2021: volume 95, issue 6, pages 809–919. DOI: 10.1111/cen.14528
- Cera G, Locantore P, Novizio R and others. ‘Pregnancy and Prenatal Management of Congenital Adrenal Hyperplasia’. Journal of Clinical Medicine 2022: volume 11, issue 20, page 6,156. DOI: 10.3390/jcm11206156
- Nowotny H, Neumann U, Tardy-Guidollet V and others. ‘Prenatal dexamethasone treatment for classic 21-hydroxylase deficiency in Europe’. European Journal of Endocrinology 2022: volume 186, issue 5, pages K17–K24. DOI: 10.1530/EJE-21-0554
- Pignatelli D, Carvalho BL, Palmeiro A and others. ‘The Complexities in Genotyping of Congenital Adrenal Hyperplasia: 21-Hydroxylase Deficiency’. Frontiers in Endocrinology 2019: volume 10. DOI: 10.3389/fendo.2019.00432
- Speiser PW, Arlt W, Auchus RJ and others. ‘Congenital Adrenal Hyperplasia Due to Steroid 21-Hydroxylase Deficiency: An Endocrine Society Clinical Practice Guideline’. The Journal of Clinical Endocrinology & Metabolism 2018: volume 103, issue 11, pages 4,043–4,088. DOI: 10.1210/jc.2018-01865
For patients
- CAH is us
- Great Ormond Street Hospital for Children NHS Foundation Trust: Congenital Adrenal Hyperplasia (PDF, three pages)
- Living with CAH