Hereditary haemochromatosis
Hereditary haemochromatosis is characterised by an excess of iron in particular organs in the body. It affects all genders equally, though women often present later than men. It is caused by pathogenic variants in the HFE gene.
Overview
Hereditary haemochromatosis is an autosomal recessive condition characterised by progressive iron overload in the body, leading to multiple organ damage and dysfunction.
Clinical features
Hereditary haemochromatosis (HH) typically presents between 30 and 60 years of age. Although both genders are equally likely to have the pathogenic genetic variants that cause HH, clinical features are more commonly seen in males. It is common within Northern European populations, especially in those with Celtic heritage. Women tend to present roughly 10 years later than men, and more commonly after menopause.
Early diagnosis is not easy, as many of the symptoms are common and non-specific. They include:
- weakness and lethargy;
- joint pain;
- weight loss; and
- loss of sex drive.
Given the non-specific nature of the early features, it is vital for clinicians to be alert to family history, particularly because early diagnosis directly improves prognosis.
Later features of HH, which are caused by excessive parenchymal storage of iron in target organs, include:
- increased skin pigmentation (bronzing);
- diabetes mellitus;
- cardiomyopathy;
- hypogonadism, leading to decreased libido, erectile dysfunction or premature menopause; and
- liver manifestations, such as:
- hepatomegaly;
- cirrhosis; and
- hepatocellular carcinoma and cholangiocarcinoma which account for one third of HH-related deaths.
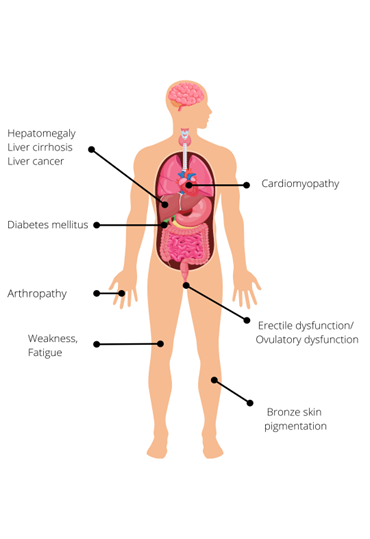
Figure 1: Clinical features of hereditary haemochromatosis.
Due to the loss of iron during menstruation, women are less likely than men to develop clinical features of iron overload. If HH is diagnosed before the development of cirrhosis or diabetes mellitus, and treated adequately by venesection, life expectancy can be normal. However, if the diagnosis is made after the onset of irreversible organ damage, life expectancy is significantly reduced, primarily due to the risk of liver cancer.
Genetics
HH is caused by pathogenic variants in the HFE gene. Pathogenic variants in this gene interfere with iron homeostasis, leading to inappropriately high iron absorption by the intestinal mucosa.
There are two common pathogenic variants that can cause HH: C282Y (p.Cys282Tyr) and H63D (p.His63Asp). Around 90% of patients with clinical HH are homozygous for C282Y (both their copies of the HFE gene have this variant), as this genotype is most at risk for iron overload. Individuals with one C282Y and one H63D pathogenic variant (compound heterozygotes) may accumulate iron, but the risk of developing clinical HH is much lower than that of C282Y homozygotes. H63D homozygotes are also far less likely to have clinical features.

Figure 2: The most common genotypes for hereditary haemochromatosis with reported allele frequencies, where red represents the C282Y variant, blue the H63D variant and green a non-pathogenic variant.
Penetrance for HH is incomplete and the condition shows variable disease expression. This means that many individuals with HH may never manifest serious clinical features. Clinical penetrance (the proportion of individuals who develop clinical features) is much less common than biochemical penetrance (the proportion of individuals with increased transferrin saturation and serum ferritin concentration but no clinical features). For this reason, population screening for the HFE genotype is not recommended.
Non-HFE HH is much rarer, being a genetically heterogeneous group of iron overload disorders caused by pathogenic variants in hemojuvelin, hepcidin, transferrin receptor 2 and ferroportin genes.
Inheritance and genomic counselling
HH is inherited in an autosomal recessive manner. The parents of most affected individuals are carriers for the condition. If both parents are carriers, with each pregnancy there is a:
- 1 in 4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected.
- 1 in 2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1 in 4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
For this reason, it is important to clarify the genetic status of the adult siblings and offspring of affected individuals in order to allow prompt initiation of treatment and preventative measures. Since the condition does not manifest before adulthood, testing children is unlikely to be valuable. Note that juvenile HH is a separate, rarer, more severe condition, with a different genetic basis. Families with HH are not generally at risk of juvenile-onset disease.
Increasingly, genomic testing for HH is being managed in primary care, with regional support from haematology services or, occasionally, clinical genetics services.
Management
Treatment of biochemical iron overload via venesection is indicated for all fit patients, with or without clinical features. The frequency of venesections may be individualised depending on the severity of symptoms, age, comorbidities and tolerability of the procedure.
Diet alone cannot treat HH, so affected individuals need not be advised extreme dietary restriction. Limiting iron fortified food or avoiding excess red meat may be helpful.
For more information, see Presentation: Patient with iron overload (suspected hereditary haemochromatosis).
Resources
For clinicians
- GeneReviews: HFE-Related hemochromatosis
- NHS England: National Genomic Test Directory
References:
- Drakesmith H, Sweetland E, Schimanski L and others. ‘The hemochromatosis protein HFE inhibits iron export from macrophages’. Proceedings of the National Academy of Sciences 2002: volume 99, issue 24, pages 15,602–15,607
- Firth HV and Hurst JA. ‘Oxford Desk Reference: Clinical Genetics and Genomics’. Oxford University Press 2017: pages 335–540. DOI: 10.1093/med/9780199557509.003.0003
- Fitzsimons EJ, Cullis JO, Thomas DW and others. ‘Diagnosis and therapy of genetic haemochromatosis (review and 2017 update)’. British Journal of Haematology 2018: volume 181, issue 3, pages 293–303. DOI: 10.1111/bjh.15164
- Pilling LC, Tamosauskaite J, Jones G and others. ‘Common conditions associated with hereditary haemochromatosis genetic variants: Cohort study in UK Biobank’. British Medical Journal 2019: volume 364, article number k5222. DOI: 10.1136/bmj.k5222
For patients
- NHS Health A to Z: Haemochromatosis: Symptoms
- British Heart Foundation: What is haemochromatosis?
- Haemochromatosis UK: What is genetic haemochromatosis?