Long QT syndrome
Long QT syndrome is characterised by QT prolongation on an electrocardiogram. It is associated with ventricular arrhythmias and sudden death, often triggered by adrenergic activation.
Clinical features
- Individuals affected by long QT syndrome (LQTS) may be asymptomatic, or may experience palpitations, dizziness and/or syncope. The first presentation may be cardiac arrest or sudden cardiac death. The mean age at presentation is 14 years.
- In LQTS type 1, arrhythmias are associated with events that result in elevated sympathetic activity, such as intense exercise and emotional stress (for example, swimming and diving). In LQTS type 2, auditory stimuli are a recognised trigger, and the postpartum period is a time of elevated risk. In patients with LQTS type 3, events tend to occur during rest or sleep, when the QT interval is further prolonged at slow heart rates.
- Congenital LQTS is diagnosed in the absence of QT prolonging medications or electrolyte anomalies (particularly hypokalaemia or hypomagnesaemia).
- LQTS diagnosis is made:
- according to the Schwartz score (risk score >3.5);
- when repeated ECGs demonstrate QTc >500ms; or
- when repeated ECGs demonstrate QTc >480ms with associated unexplained or likely arrhythmic syncope (see figure 1).
- Lying and standing ECGs or exercise stress testing can be used to unmask latent QT prolongation, as the QT will fail to shorten at increased heart rates and in early recovery.
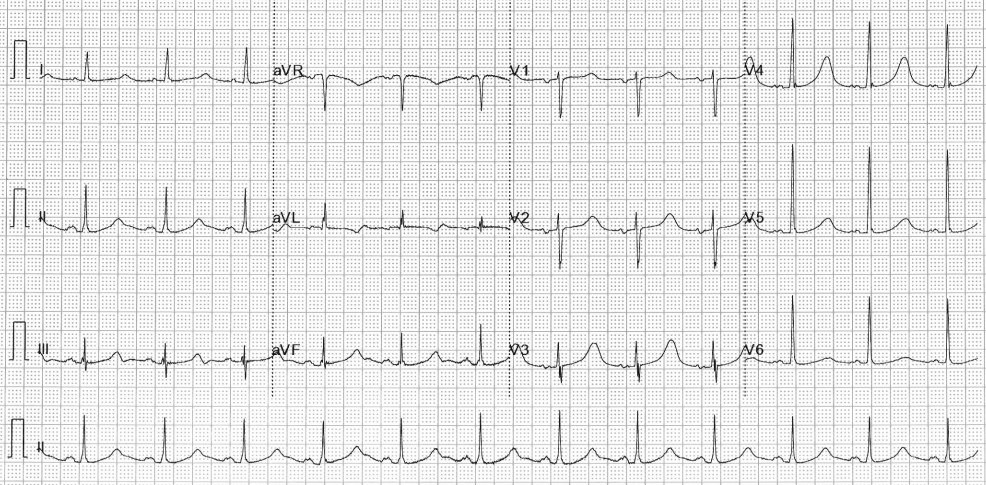
Figure 1: ECG demonstrating QT prolongation in a patient with Jervell and Lange-Nielson syndrome
Genetics
- There are numerous subtypes of LQTS, defined by the gene containing the pathogenic variant.
- Approximately 75% of those with a clinical diagnosis of LQTS have a variant in one of the LQTS associated genes: KCNQ1 (LQTS type 1), KCNH2 (LQTS type 2) and SCN5A (LQTS type 3). These genes account for 90% of variants with an autosomal dominant pattern of inheritance.
- Some subtypes of LQTS are associated with additional phenotypes. Examples include:
-
- Andersen-Tawil syndrome (caused by variants in KCNJ2, inherited in an autosomal dominant pattern), also known as LQTS type 7, which is associated with a prolonged QT interval, frequent (often bidirectional) ventricular arrhythmias, periodic paralysis and dysmorphic facial features; and
-
- Jervell and Lange-Nielson syndrome (caused by biallelic variants in KCNQ1 or KCNE1, inherited in an autosomal recessive pattern), in which QT prolongation is associated with sensorineural hearing loss.
Inheritance and genomic counselling
- LQTS is most commonly inherited in an autosomal dominant pattern, which means that there is a 50% chance of an affected parent passing the condition to each child. However, recessive forms of the condition do exist.
- Taking a three-generation family history and identifying any previously known familial pathogenic variants is important for genomic counselling. Between and within families, there is variability in disease severity, likely due to the presence of ‘modifier’ genes that increase or decrease the impact of a pathogenic variant.
Management
- All patients with LQTS should be given lifestyle advice.
- Certain drugs may increase the likelihood of arrhythmia in the setting of LQTS, and patients should be directed to CredibleMeds for a list of drugs to avoid.
- During a diarrhoea and vomiting illness, affected individuals should keep well hydrated using oral rehydration therapy (such as Dioralyte).
- Affected individuals should avoid gene-specific triggers (such as competitive swimming in LQTS type 1).
- The mainstay of treatment is beta blockade (ideally non-selective beta blockers such as nadolol or propranolol).
- Mexiletine is indicated for patients with LQTS type 3 and QT prolongation.
- Flecainide and/or acetazolamide should be considered in patients with LQTS type 7.
- Implantable cardioverter-defibrillator (ICD) implantation is recommended in individuals who have survived cardiac arrest, or have arrhythmic syncope or haemodynamically non-tolerated ventricular arrhythmia despite beta blockade and genotype-specific therapy.
- Left sympathetic cardiac denervation (LSCD) is indicated in symptomatic individuals who decline ICD therapy or who have arrhythmic syncope or haemodynamically non-tolerated ventricular arrhythmia despite beta blockade and genotype-specific therapy (with ICD in situ).
- ICD implantation or LCSD should be considered in patients with symptomatic LQTS when beta-blockers and genotype-specific therapies are not tolerated or contraindicated at the therapeutic dose.
Resources
For clinicians
- NHS England: National Genomic Test Directory
References:
- Zeppenfeld K, Tfelt-Hansen J, de Riva M and others. ‘2022 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden death of the European Society of Cardiology (ESC) endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC)‘. European Heart Journal 2022: volume 43, issue 40, pages 3,997–4,126. DOI: 10.1093/eurheartj/ehac262
For patients
- British Heart Foundation: Long QT syndrome
- Cardiac Risk in the Young
- CredibleMeds
- NHS Health A to Z: Long QT syndrome