Multiple endocrine neoplasia type 4
Multiple endocrine neoplasia type 4 is a condition associated with variants in the CDKN1B gene that causes predisposition to tumours in the endocrine organs.
Overview
Multiple endocrine neoplasia type 4 (MEN4) is a condition that causes predisposition to tumours in the endocrine organs. It is caused by constitutional (germline) pathogenic variants of the CDKN1B gene.
Clinical features
Clinical features of MEN4 include:
- parathyroid tumours;
- pituitary tumours; and
- endocrine tumours of the gastro-entero-pancreatic tract – see figure 1.
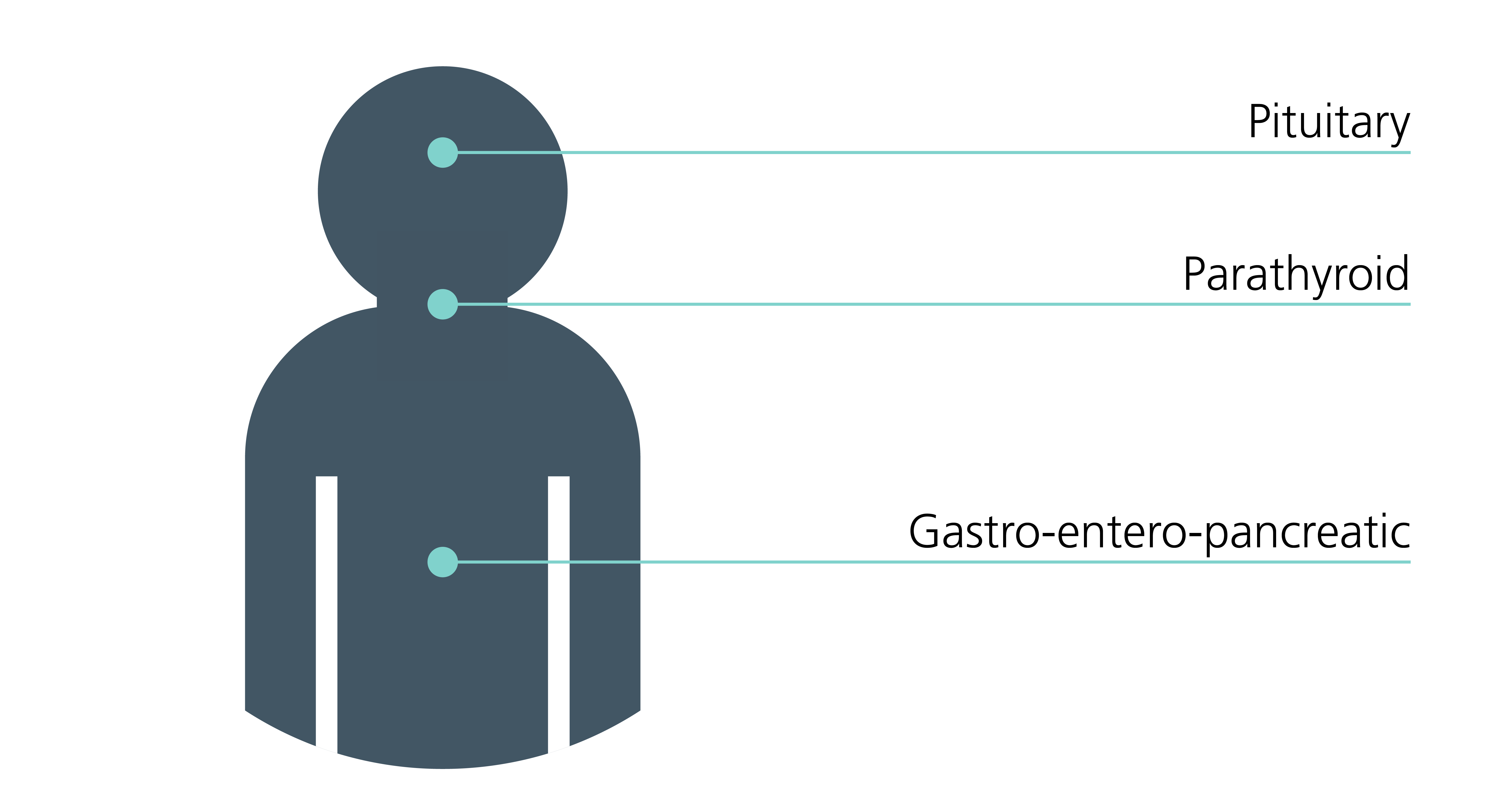
Figure 1: Sites of endocrine tumours associated with MEN4
Genetics
MEN4 is caused by constitutional (germline) pathogenic loss-of-function variants in the CDKN1B tumour suppressor gene, which encodes for the cell cycle inhibitor p27 protein.
MEN4 has a similar phenotype to MEN1 and is thought to account for a little more than 1% of individuals with an MEN1 phenotype. In rare instances, it has also been identified in children with corticotroph tumours. Due to its rarity, it is difficult to ascertain whether tumours of non-endocrine organs may be a component of MEN4. There is no clear genotype-phenotype correlation. Rarely, pathogenic variants have also been described with other cell cycle inhibitors (CDKN1A, CDKN2B or CDKN2C); however, NHS-funded testing of these genes will not be recommended until further data regarding disease association and penetrance are available.
For information about genomic testing, see Presentation: Patient with endocrine neoplasia and Presentation: Patient with possible multiple endocrine neoplasia type 1.
Inheritance and genomic counselling
MEN4 is an autosomal dominant condition, although penetrance is difficult to estimate due to its rarity. A parent with a pathogenic variant has a 50% chance of passing the variant on to their child.
Family planning implications
The Human Fertilisation & Embryology Authority has approved the use of preimplantation genetic testing for monogenic disorders (PGT-M) (previously known as pre-implantation genetic diagnosis) for couples in whom one potential parent is a carrier of a pathogenic variant in MEN1. It is best practice that discussions regarding PGT-M and other family planning options be undertaken by a specialist genetic counsellor or a clinical geneticist.
Alternative options may include:
- prenatal testing (invasive, or non-invasive if the intended father is the carrier) with termination of affected embryos;
- adoption;
- gamete donation; or
- natural conception and pregnancy with testing of children in adulthood.
Management
Management and screening of individuals with MEN4 should follow published MEN1 guidelines (see table 1 and the resources section below). Endocrine neoplasia syndromes are multisystem conditions and care is best delivered by a multidisciplinary team.
Table 1: Screening in individuals at high risk of developing MEN1
| Tumour | Age to begin screening (years) | Annual biochemical test (plasma or serum) | Imaging test (time interval) |
| Parathyroid | Eight | Calcium, parathyroid hormone (PTH) | None |
| Gastrinoma | 20 | Gastrin (with or without gastric pH) | None |
| Insulinoma | Five | Fasting glucose, insulin | None |
| Other pancreatic neuroendocrine tumours | Under 10 | Chromogranin-A, pancreatic polypeptide, glucagon, VIP | MRI (preferably in children), CT or EUS (annually) |
| Anterior pituitary | Five | Prolactin, insulin-like growth factor 1 (IGF-1) | MRI (every three years) |
| Adrenal | Under 10 | None, unless symptoms or signs of functioning tumour and/or tumour under 1cm are identified on imaging | MRI (preferably in children) or CT (annually; simultaneous with pancreatic imaging) |
| Thymic and bronchial carcinoid | 15 | None | MRI (preferably in children) or CT (every one to two years) |
Resources
For clinicians
- Genomics England: Genomic Medicine Service (GMS) Signed Off Panels Resource
- NHS England: National Genomic Test Directory
References:
- Thakker RV, Newey PJ, Walls GV and others. ‘Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1)’. Journal of Clinical Endocrinology & Metabolism 2012: volume 97, issue 9, pages 2,990–3,011. DOI: 10.1210/jc.2012-1230