Polycystic liver disease
The majority of patients with polycystic liver disease (PLD) are asymptomatic. In some cases, however, cyst burden can significantly impact quality of life, necessitating intervention. PLD is characterised by multiple (>10) cysts within the liver parenchyma.
Overview
Multiple liver cysts may be seen in autosomal dominant polycystic kidney disease (ADPKD), in which multiple kidney cysts are also present, or in autosomal dominant polycystic liver disease (ADPLD), in which cysts are mainly restricted to the liver. ADPLD has been linked to pathogenic variants in multiple different genes, with 20%–40% of cases linked to variants in the PRKCSH and SEC63 genes. Patients can present at any age, but typically in the third decade. Most patients remain asymptomatic and monitoring is guided by symptom burden. Medical and surgical treatment options can be considered; however, the only definitive treatment, used for the most severe cases only, is liver transplantation.
Clinical features
Many patients with ADPLD remain asymptomatic. In those who do develop symptoms, these are predominantly related to cyst burden and can range from very mild to severe symptoms that significantly impact quality of life.
Cyst growth and accumulation can cause hepatomegaly (enlargement of the liver) and compression of surrounding anatomical structures. Clinical features include:
- abdominal pain;
- back pain;
- early satiety;
- shortness of breath;
- malnutrition;
- dyspepsia/gastro-oesophageal reflux disease; and
- limited mobility.
Rarely, progressive liver cysts can cause structural complications affecting liver function. Clinical features include:
- jaundice caused by the compression of the biliary tree by cysts; and
- ascites and other features of portal hypertension caused by compression of the hepatic vasculature by cysts (note that this presentation could also be due to concomitant congenital hepatic fibrosis).
Cyst complications are rare and include:
- cyst haemorrhage (bleeding into a cyst);
- cyst infection (presenting with pain and fever); and
- cyst rupture (presenting with severe abdominal pain).
Genomics
ADPLD is a disease characterised by the progressive development of numerous liver cysts in adulthood due to embryonic ductal plate malformation.
It is an autosomal dominant disorder that has been linked to pathogenic variants in a number of different genes. About 20% of cases are caused by pathogenic variants in the PRKCSH gene, and about 15% of cases by pathogenic variants in the SEC63 gene. Both of these lead to errors in glycoprotein synthesis which are associated with embryological malformations contributing to liver cyst formation.
Pathogenic variants linked to ADPLD have also been identified in the ALG8, GANAB, LRP5, PKHD, ALR9 and SEC61B genes. However, as in more than 50% of ADPLD cases a pathogenic variant is not identified, it is likely that other genes are also involved. This genetic heterogeneity likely contributes to the spectrum of disease severity.
ADPLD is estimated to affect about 1 in 10,000 people. This is likely to be an underestimate as many patients are asymptomatic.
Diagnosis
ADPLD is diagnosed in the presence of >10 liver cysts on imaging.
- Imaging features of the cysts seen in ADPLD are comparable with simple liver cysts.
- In the presence of symptoms, MRI (see figure 1) is the preferred imaging modality to:
- identify smaller cysts;
- evaluate cyst distribution in the liver, including the volume of unaffected liver remaining; and
- ascertain the degree of hepatic vasculature or biliary tree compression.
- No further investigation is required if ultrasound or CT are suggestive of ADPLD in the absence of symptoms.
- There are a few classification systems used to stratify ADPLD that consider number, size and distribution of hepatic cysts.
Genomic testing to look for pathogenic variants in the genes that are recognised to cause ADPLD is available through the National Genomic Test Directory. For instance, the R173 Polycystic liver disease clinical indication is a gene panel that includes assessment for frequently seen pathogenic variants. This is a particularly relevant diagnostic test in those with a positive family history. For more information about genomic testing for ADPLD, see Presentation: Patient with polycystic liver disease.
Blood results are usually normal; however, in some cases alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) may be raised. In rare cases, a raised bilirubin level can indicate compression of the biliary tree and a low platelet count can indicate compression of the portal vasculature.
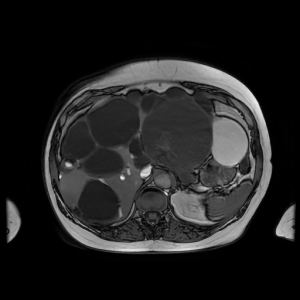
Figure 1: MRI scan of a patient with polycystic liver disease. This cross-sectional image of a polycystic liver shows liver enlargement due to multiple cysts of varying size (shown as dark saccular areas). Some liver parenchyma is visible but there is gross replacement by cysts here. Image provided by Dr Bill Griffiths and with consent from the patient.
Inheritance and genomic counselling
ADPLD is a disorder with autosomal dominant inheritance and therefore most patients with it have a positive family history. In some cases, however, de novo genetic variants can occur, which can manifest as ADPLD without a positive family history.
For patients with ADPLD with a positive family history, the gene with the pathogenic variant could have been inherited from either parent. Genetic counselling is recommended for patients, including pre-conception counselling regarding risk of passing on the disease.
- Individuals affected by an autosomal dominant condition have one working copy of the gene, and one with a pathogenic variant.
- The chance of a child inheriting the gene with the variant from an affected parent is 1 in 2 (50%).
- Incomplete penetrance can occur when not everyone who has the variant develops the disease.
The risk of developing ADPLD is equal for males and females; however, females are at greater risk of developing more severe presentation of the disease.
Routine genetic screening for family members is not recommended due to the variability of the condition. The more useful test to determine an affected individual – if felt necessary, as there is no treatment for asymptomatic disease – would be a liver ultrasonography.
Management
The management of ADPLD is primarily focused on addressing symptom burden by reducing liver volume in symptomatic patients. Treatment options should be considered on a case-by-case basis. Asymptomatic patients do not require treatment or monitoring.
Somatostatin analogies, including octreotide, can be used as medical management to slow cyst growth and fluid secretion in the liver. The limited long-term benefit and side-effect profile should be considered.
Interventional and surgical management options include:
- percutaneous aspiration sclerotherapy (interventional radiology), as recurrence is common;
- cyst fenestration (de-roofing), which is effective for symptom control and long-term reduction in cyst volume but has no impact on natural progression of the disease;
- segmental hepatic resection, resulting in substantial liver volume reduction and effective symptom control but with significant comorbidity association, so only considered if liver transplantation is not an option; and
- liver transplantation, which is the only definitive curative treatment option.
Considering risk factors:
- exogenous oestrogen use should be avoided following diagnosis of ADPLD, as it can lead to accelerated cyst growth;
- multiple pregnancies; and
- female sex.
There are currently no approved gene therapies for ADPLD, and there is a need to develop novel therapeutic strategies targeting cyst growth.
Resources
For clinicians
- Genomics England’s PanelApp: Polycystic liver disease
- National Organization for Rare Disorders: Polycystic Liver Disease
References:
- European Association for the Study of the Liver. ‘EASL Clinical Practice Guidelines on the management of cystic liver diseases‘. 2022 Journal of Hepatology: volume 77, issue 4, pages 1,083–1,108. DOI: 10.1111/liv.14349
- Olaizola P, Rodrigues PM, Caballero-Camino FJ and others. ‘Genetics, pathobiology and therapeutic opportunities of polycystic liver disease‘. Nature Reviews Gastroenterology & Hepatology 2022: volume 19, issue 9, pages 585–604. DOI: 10.1038/s41575-022-00617-7
- Yu Z, Shen X, Hu C and others. ‘Molecular Mechanisms of Isolated Polycystic Liver Diseases‘. Frontiers in Genetics 2022: volume 13, articles number 846,877. DOI: 10.3389/fgene.2022.846877
For patients
- British Liver Trust: Polycystic liver disease