Short QT syndrome
Short QT syndrome is a rare inherited arrhythmia syndrome characterised by an abnormally short QT interval, which increases the risk of cardiac arrhythmias and sudden cardiac death. It is caused by pathogenic genetic variants that alter the function of ion channels responsible for currents that generate the cardiac action potential.
Clinical features
- Individuals with short QT syndrome (SQTS) may present with palpitations, syncope or cardiac arrest. Syncope without warning is suspicious for underlying ventricular arrhythmia. Cardiac arrest is common, and SQTS appears to have high rates of lethality in all age groups.
- The condition is associated with a short QT interval, premature atrial fibrillation and ventricular fibrillation in the absence of structural heart disease.
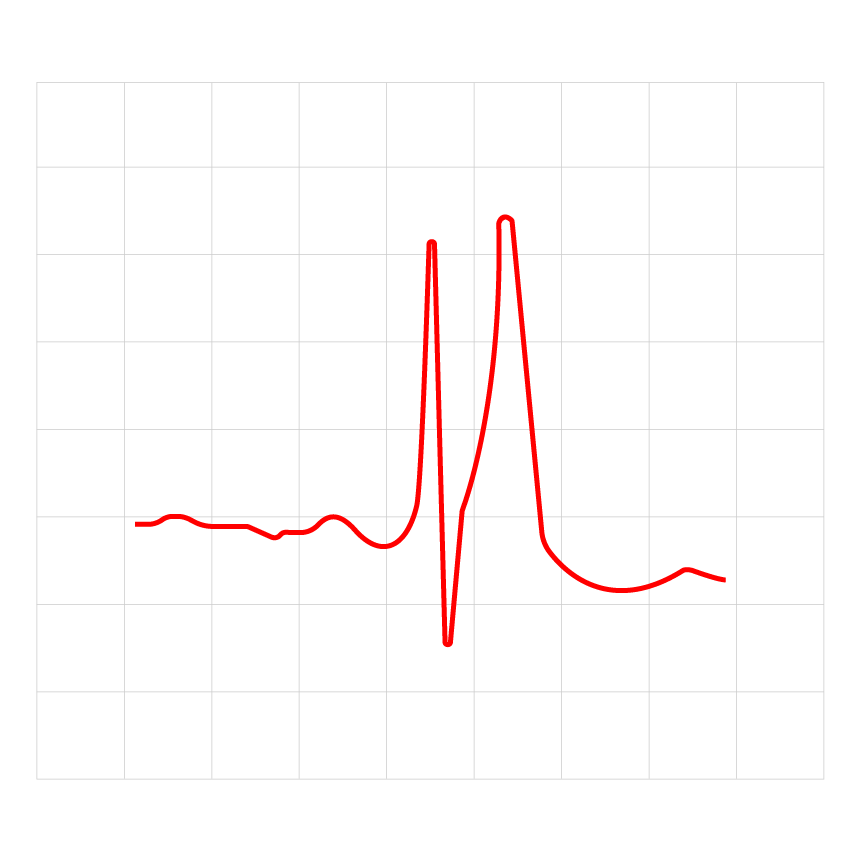
Figure 1: The ECG result of an adult patient with short QT syndrome
Genetics
- The four genes in table 1 below have been implicated in SQTS, and are tested for in the National Genomic Test Directory clinical indication R130, which involves a small gene panel test.
| Type of SQT | Gene | Current | Phenotype |
| SQT1 | KCNH2 | IKr |  |
| SQT2 | KCNQ1 | IKs |  |
| SQT3 | KCNJ2 | IK1 |  |
| SQT4 | CACNA1C | ICaL | 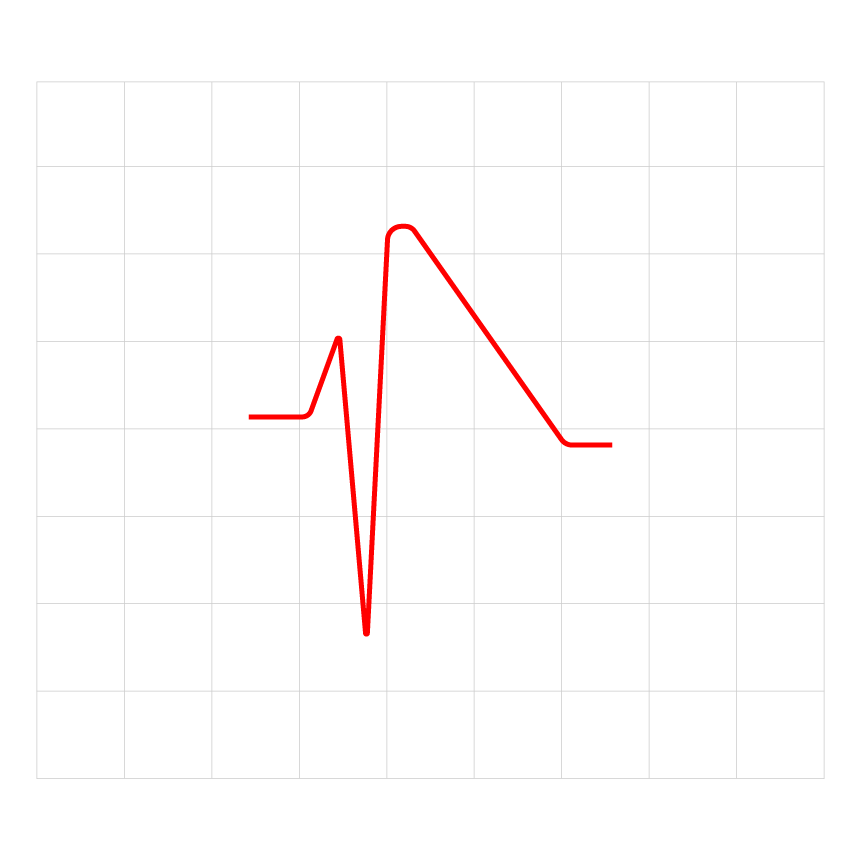 |
| SQT5 | CACNB2B | ICaL | 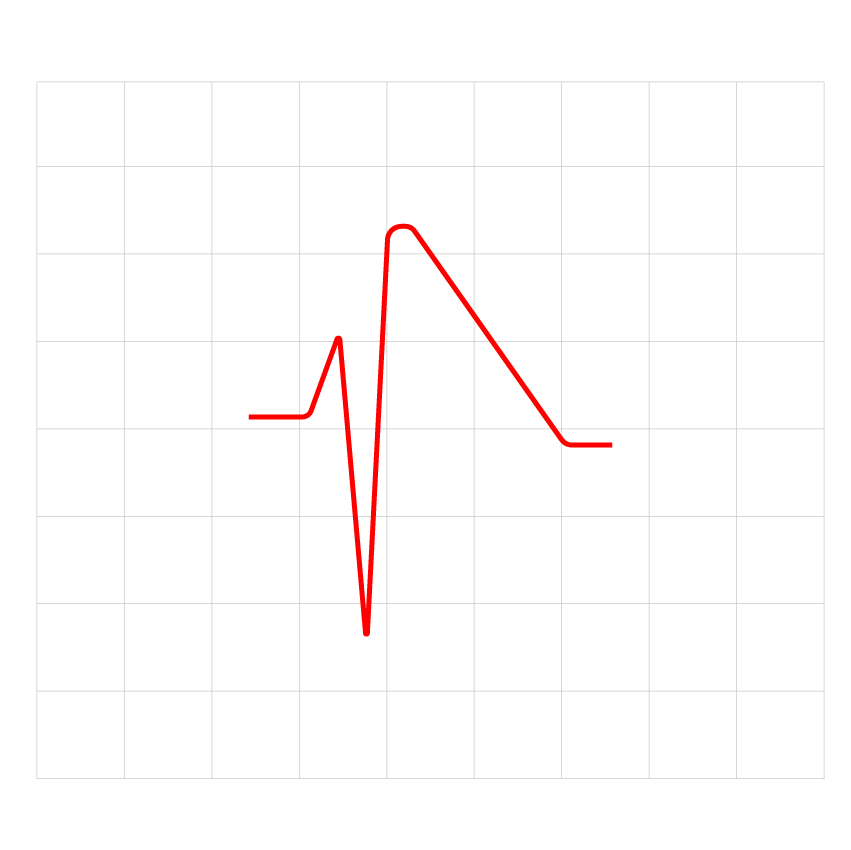 |
Table 1: The genes associated with SQT
- SQTS genotypes one to three are produced by gain-of-function pathogenic variants in myocardial potassium channels. Interestingly, loss-of-function pathogenic variants in the same genes are implicated in long QT syndrome.
- SQTS genotype four is produced by loss-of-function pathogenic variants in the L-type cardiac channel.
- About 15% of SQTS cases are associated with variants in KCNH2. Missense variants in KCNJ2, KCNQ1 and CACNA1C have been found in a handful of familial cases.
Inheritance and genomic counselling
- Although de novo cases of SQTS have been reported, the condition usually has an autosomal dominant pattern of inheritance, which means that there is a 50% chance of affected parents passing the condition on to each child. Taking a three-generation family history and identifying any previously known familial pathogenic variants is important to facilitate genomic counselling.
- The testing criteria for SQTS can be found in the National Genomic Test Directory. For more information about testing, see Adult with short QT syndrome.
- All first-degree family members of an affected individual should be offered clinical screening. Where the pathogenic genetic variant is known, other family members should be offered predictive testing (as part of cascade testing).
Management
- Affected individuals should avoid medications that shorten the QT interval (such as nicorandil).
- Implantable cardioverter-defibrillator (ICD) implantation is recommended in SQTS patients who have survived cardiac arrest or have had documented episodes of spontaneous sustained ventricular arrhythmia.
- ICD implantation should be considered in SQTS patients who have arrhythmic syncope.
- Close monitoring with insertion of an implantable loop recorder should be considered in young children with SQTS.
- Pharmacological therapy (such as quinidine) may be considered in the following situations:
-
- in patients with contraindications to ICD implantation or who decline ICD implantation;
-
- as an adjunct therapy to prevent appropriate ICD discharges for ventricular arrhythmias;
-
- to treat episodes of atrial fibrillation;
-
- as an alternative to ICD implantation in young children; and
-
- in young patients who are asymptomatic but have a family history of sudden cardiac death.
Resources
For clinicians
- Life in the Fast Lane: Short QT syndrome
- NHS England: National Genomic Test Directory
References:
- Zeppendelf K, Tfelt-Hansen J, de Riva M and others. ‘2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: Developed by the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC) endorsed by the Association for European Paediatric and Congenital Cardiology (AEPC)‘. European Heart Journal 2022: volume 43, issue 40, pages 3,997–4,126. DOI: 10.1093/eurheartj/ehac262
For patients
- Cardiac Risk in the Young: Short QT syndrome
- myheart: Short QT syndrome