Spinal muscular atrophy
Spinal muscular atrophy is a genetic condition characterised by weakness and wasting in the skeletal muscles, which causes problems with movement.
Overview
The muscle weakness and hypotonia characteristic of spinal muscular atrophy (SMA) is caused by loss of anterior horn cells in the spinal cord (lower motor neurons) and the brain stem nuclei. Cognitive function is normal.
Clinical features
Clinical features of SMA include:
- progressive muscle weakness: proximal muscles are usually more severely affected than distal muscles;
- hypotonia;
- areflexia or hyporeflexia; and
- tongue fasciculations.
There are different types of SMA, which are characterised by the age of onset and the severity of symptoms. Each type, including age of onset, presentation and prognosis, is listed below.
- SMA type 0:
- prenatal onset;
- often, reduced fetal movements will have been identified; postnatally there will be respiratory failure at birth, severe weakness, absent reflexes and arthrogryposis, and
- most babies will not live beyond six months.
- SMA type 1 (also known as Werdnig-Hoffman disease):
- age of onset is under 6 months (mean age of onset is two and a half months);
- babies may manage to develop some head control but will have a progressive muscular weakness and are therefore unlikely to sit unsupported, and there may be sucking and/or swallowing difficulties; and
- the median age of survival is 8 to 10 months.
- SMA type 2:
- age of onset is 6 to 18 months;
- there will be proximal muscle weakness and delayed developmental milestones, with loss of some skills, as well as reduced or absent reflexes; and
- most survive into adulthood.
- SMA type 3:
- onset can be any time during childhood (over 18 months);
- patients will achieve normal ambulation but will experience progressive difficulties running and climbing; loss of motor skills and fatigue are common; and
- there is a normal life expectancy.
- SMA type 4:
- onset can be any time during adulthood;
- there will be fatigue and proximal muscle weakness; and
- there is a normal life expectancy.
Genomics
SMA is caused by loss of both copies of the SMN1 gene (most frequently deletions of both gene copies).
Genomic testing checks how many copies of the SMN1 gene an individual has. Someone with no copies has SMA, while an individual with one copy, or two copies on one chromosome and none on the other, is a carrier.
There are three points to remember when it comes to genomic testing for SMA.
- The variability of disease severity is affected by the number of copies of the SMN2 gene: another gene that is able to produce small quantities of functional SMN protein. Individuals can have between one and eight copies of SMN2. A baby with SMA type 0 is likely to have only one copy of SMN2, whereas an individual with SMA type 4 is likely to have four or more copies.
- Some carriers of SMA have two functional copies of SMN1 on one chromosome (in cis) and a deletion of SMN1 on the homologous chromosome (in trans) – see figure 1. This occurs in 5%–8% of SMA carriers and is known as the 2+0 configuration. These individuals will not be identified as carriers by standard genomic testing methods, which only identify an individual as a carrier if they have fewer than two copies of the SMN1 gene. Their offspring may inherit the chromosome without a copy of the SMN1 gene, in which case they will either be a carrier or will have SMA, depending on if they inherit a copy from their other parent.
- In 2% of individuals with SMA, one SMN1 deletion is acquired de novo, which means that only one parent is a carrier.
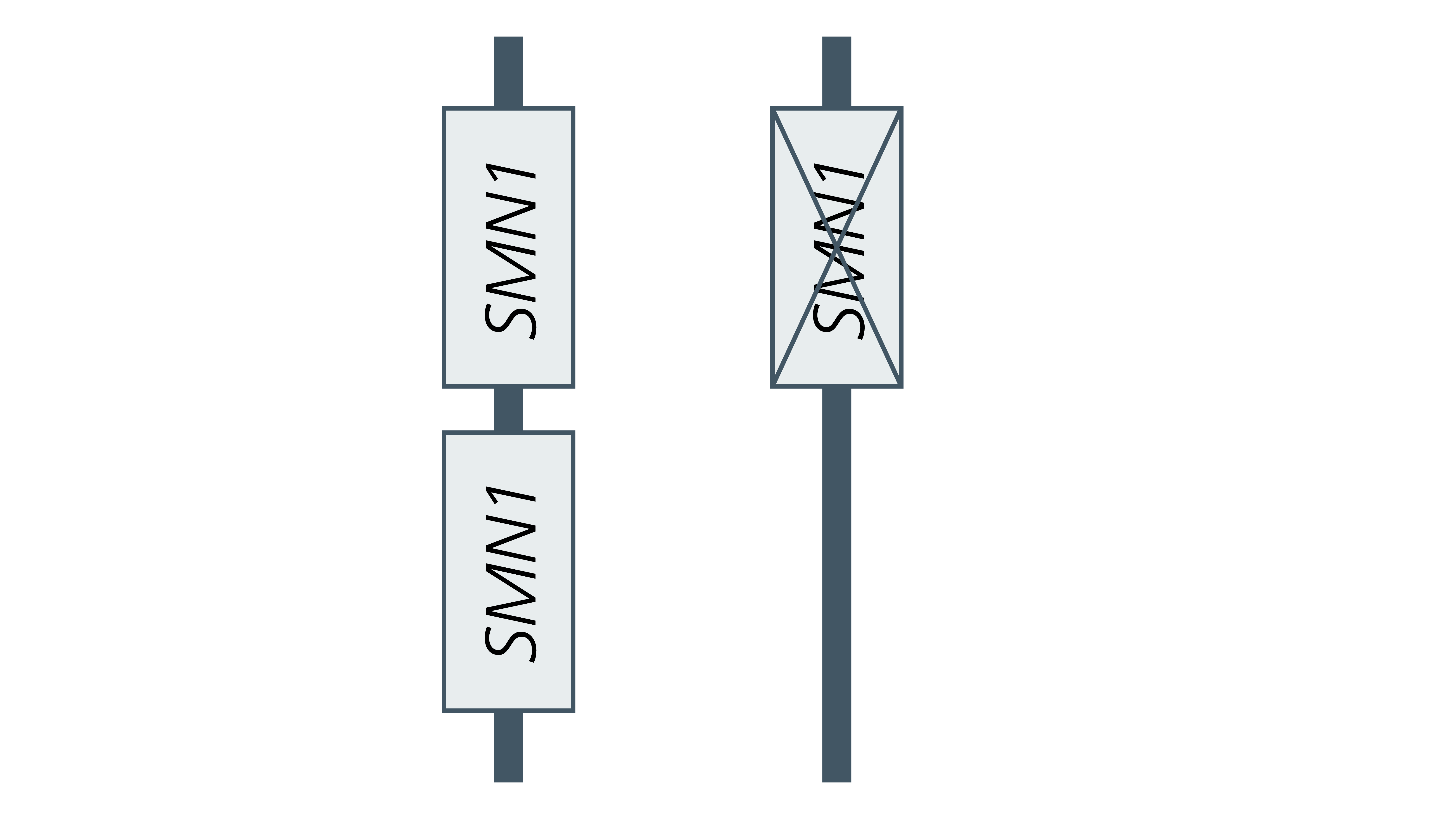
Figure 1: The 2+0 configuration
For information about testing, see Presentation: Hypotonic infant.
Diagnosis
Diagnosis is based on clinical features, confirmed by genomic testing. For information about testing, see:
Inheritance and genomic counselling
SMA is an autosomal recessive condition.
- If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
- 1 in 4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected.
- 1 in 2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1 in 4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
For most affected individuals, the parents are carriers for the condition. Therefore they have a 25% (one in four) chance of having another affected child.
Prenatal testing for SMA may be considered in a pregnancy with a known family history in either parent, or when both parents are known to be carriers for SMA. Preimplantation genetic testing may also be an option. For more information, see Presentation: Pregnancy at risk of spinal muscular atrophy.
Management
Management of children with SMA is complex and should be delivered via a multidisciplinary team, with detailed suggested approaches published by several authors (see our resources list below).
Gene-directed therapies and trials
- Nusinersen (Spinraza): This is an antisense oligonucleotide that allows the body to produce more and better-quality (longer-length) SMN from the SMN2 gene.
- Onasemnogene abeparvovec-xioi (Zolgensma): Using a vector, the faulty SMN1 gene is replaced with a working copy.
Resources
For clinicians
- Genomics England: NHS Genomic Medicine Service (GMS) Signed Off Panels Resource
- NHS England: National Genomic Test Directory
- NICE guidance: Nusinersen for treating spinal muscular atrophy
- NICE guidance: Onasemnogene aveparvovec for treating spinal muscular atrophy
- SMA News Today: Spinraza (Nusinersen) for SMA
- U.S. National Library of Medicine: ClinicalTrials.gov database
References:
- Mercuri E, Finkel RS, Muntoni F and others. ‘Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care’. Neuromuscular Disorders 2018: volume 28, issue 2, pages 103–115. DOI: 10.1016/j.nmd.2017.11.005
- Mercuri E, Finkel RS, Meyes OH and others. ‘Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics’. Neuromuscular Disorders 2018: volume 28, issue 3, pages 197–207. DOI: 10.1016/j.nmd.2017.11.004
For patients
- NHS Health A to Z: Spinal muscular atrophy
- Spinal Muscular Atrophy UK