Thalassaemia
Thalassaemia is the name of a group of genetic conditions that affect haemoglobin production and, as a result, red blood cell size.
Overview
The severity of thalassaemia varies, from patients who are completely asymptomatic in the carrier state to patients who are severely affected and require regular blood transfusions. It can be a cause of severe anaemia in utero (fetal hydrops). There are two primary groups of thalassaemia – alpha and beta – named after the type of haemoglobin chain that is affected in each group.
Clinical features and genetics
Alpha thalassaemia
Clinical features depend upon how many of the four alpha (𝞪) globin genes are involved.
As there are four alleles in total (two each for HBA1 and HBA2, both located on the short arm of chromosome 16), alpha thalassaemia has four phenotypes. These are listed below, along with the associated clinical features.
- One allele affected (alpha thalassaemia silent): The affected individual will have no symptoms.
- Two alleles affected (alpha thalassaemia trait): The affected individual will have mild microcytic and hypochromic anaemia. This can occur as alpha plus (𝞪 – / 𝞪 -) or alpha zero (- – / 𝞪 𝞪).
- Three alleles affected (haemoglobin H (HbH) disease): The affected individual will have microcytic, hypochromic anaemia with target cells and Heinz bodies, and may have hepatosplenomegaly.
- Four alleles affected (alpha thalassaemia major, also known as haemoglobin Bart syndrome – the most severe form of alpha thalassaemia): Without stem cell transplant or repeated blood transfusion, fetal hydrops and stillbirth or early neonatal death will occur.
Patients with one or two gene deletions will have minor anomalies of the full blood count, but they will be completely asymptomatic. It is important that they are not given empiric iron therapy on the basis of red blood cell size alone; ferritin should be used to assess their iron status.
Alpha thalassaemia trait can be identified from the full blood count and a normal HbA2 level on Hb electrophoresis, and genomic testing is not mandated in childhood. However, the diagnosis does have implications when the individual is planning their own family, and testing of the partner may be indicated together with genomic testing to confirm the genotype.
Patients with three affected alpha globin genes (HbH disease) tend to follow a variable course, with some requiring transfusions intermittently. Severely affected children, if untreated, can have the following symptoms and/or signs and may require a transfusion programme:
- symptoms of anaemia: tiredness, pallor, palpitations, shortness of breath and resultant poor growth;
- gallstones and/or jaundice (due to increased red blood cell destruction);
- extramedullary haematopoiesis (blood formation outside of the bone marrow) resulting in signs such as frontal bossing (a pronounced appearance of the forehead) and splenomegaly; and
- high blood pressure in pregnancy.
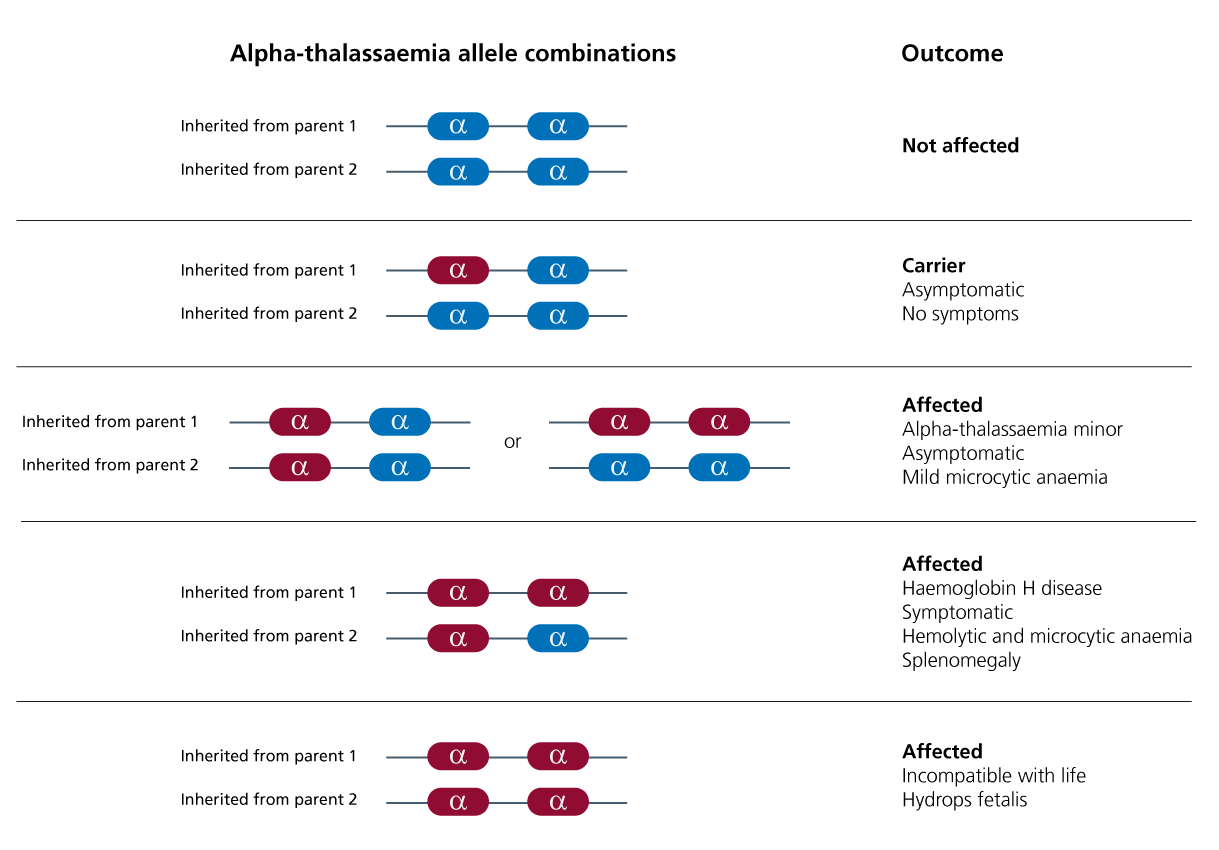
Figure 1: Alpha thalassaemia genetics and clinical consequences
Note: Red alleles have been deleted and blue alleles are present.
Beta thalassaemia
Although only one gene (the beta (𝞫) globin gene, HBB, on chromosome 11) is involved in the pathogenesis of beta thalassaemia, the type of variant greatly dictates the phenotype. Variants resulting in an absence of protein production are more severe (denoted as the 𝞫o allotype) than variants allowing some 𝞫-chain formation (𝞫+ allotype) or normal levels of 𝞫 globin with reduced function (𝞫 allotype). As such, three clinical phenotypes have been defined and are listed below.
- Beta thalassaemia minor (genotypes are 𝞫+/𝞫 and 𝞫o/𝞫): The affected individual will have mild microcytic anaemia.
- Beta thalassaemia intermedia (genotypes are 𝞫+/𝞫+ and 𝞫o/𝞫+): The affected individual will have moderate microcytic anaemia that may necessitate transfusions and may also result in the symptoms described below.
- Beta thalassaemia major (genotype is 𝞫o/𝞫o): The affected individual will have severe microcytic, hypochromic anaemia with splenomegaly, severe bone deformities and iron overload.
Children with beta thalassaemia minor have no symptoms or signs, and the diagnosis is of no clinical significance to the health of the child. However, it is important that they are not given empiric iron therapy on the basis of red blood cell size alone; ferritin should be used to assess their iron status.
Diagnosis of beta thalassaemia trait can be made on the basis of an elevated HbA2 level on Hb electrophoresis. As is the case with alpha thalassemia trait, the diagnosis has potential implications when the individual is planning their own family and partner testing may be indicated.
Children with thalassaemia intermedia can follow a variable course, with some having significant symptoms and needing regular transfusion.
Children with thalassemia major require a regular transfusion programme to prevent complications and facilitate normal growth and development. Symptoms and signs of untreated thalassaemia major include:
- symptoms of anaemia: tiredness, pallor, palpitations, shortness of breath and resultant poor growth;
- extramedullary haematopoiesis (blood formation outside of the bone marrow) resulting in signs such as frontal bossing (a pronounced appearance of the forehead), maxillary hypertrophy resulting in exposure of the upper teeth, and splenomegaly;
- gallstones and/or jaundice (due to increased red blood cell destruction); and
- children with thalassaemia can develop iron overload, resulting in liver, heart and/or hormonal problems, and those on a transfusion programme will require regular iron chelation therapy (medication to remove excess iron from the body).
For information about testing, see Presentation: Child with anaemia.
Inheritance and genomic counselling
Alpha thalassaemia
Alpha thalassaemia is inherited in an autosomal recessive pattern; however, due to the involvement of two genes, inheritance is complex and entirely dependent on the parents’ specific allotypes. If the parents both have the alpha-zero genotypes, one in four of their pregnancies will result in alpha thalassaemia major.
Beta thalassaemia
Beta thalassaemia is also inherited in an autosomal recessive manner. However, due to the variable clinical effect of the genotypes, this inheritance can also be complex. For example, if the parents are both of the 𝞫o/𝞫 genotypes, they have a one-in-four chance of having a child with beta thalassaemia major. However, if they are of the 𝞫+/𝞫 genotype, their offspring is extremely unlikely (barring de novo variants) to have beta thalassaemia major.
There is a national haemoglobinopathy screening programme in the UK which will identify potentially affected women during pregnancy and will trigger partner testing and referral to haematology. Some families may choose to have partner testing when planning a pregnancy.
Management
Management of children with thalassaemia is complex and should be delivered via a multidisciplinary team in a haemoglobinopathy specialist centre. There are national standards of clinical care for children with thalassaemia.
Targeted gene therapy for the management of thalassaemia is moving from clinical trials to clinical practice (see reference list below).
Resources
For clinicians
- British Society for Haematology: Significant haemoglobinopathies: Guidelines for screening and diagnosis
- Genomics England: NHS Genomic Medicine Service (GMS) Signed Off Panels Resource
- NHS England: National Genomic Test Directory
- Orphanet: Alpha-thalassemia
- Orphanet: Beta-thalassemia
- Patient info: Thalassaemia
- US National Library of Medicine: ClinicalTrials.gov database
References:
- Payen E. ‘Efficacy and safety of gene therapy for β-thalassemia‘. The New England Journal of Medicine 2022: volume 386, issue 5, pages 488–49. DOI: 10.1056/NEJMe2118580
- Thompson AA, Walters MC, Kwiatkowski J and others. ‘Gene therapy in patients with transfusion-dependent β-thalassemia’. The New England Journal of Medicine 2018: volume 378, issue 16, pages 1,479–1,493. DOI: 10.1056/NEJMoa1705342
For patients
- NHS Health A to Z: Thalassaemia
- Patient info: Thalassaemia
- UK Thalassaemia Society